
Par le Pr Jean-Michel Claverie
Dans plusieurs interviews récentes, le Pr Luc Montagnier a déclaré que le SARS-CoV-2 serait un virus manipulé par les Chinois et qui contiendrait de l'ADN de VIH (le virus du SIDA) ! Cette allégation reposerait sur la détection de similarités entre le génome du SARS-CoV-2 et celui du VIH.
Des séquences de « bases » désignées par des lettres
Les « textes » de ces génomes, écrits en une suite de « bases », désignées par des lettres (A, T, G, C), pour les deux virus, sont publiquement accessibles dans les bases de données publiques comme celle du National Institute for Biotechnology Information aux Etats-Unis.
Ces deux génomes, longs de 30.000 lettres (ou bases) pour le SARS-CoV-2 et de 9.200 lettres (ou bases) pour le VIH, codent, entre autres, pour les protéines qui permettent aux virus de se multiplier et de fabriquer les particules virales qui permettent leur dissémination.
Comment détecter une manipulation génétique ?
Grace aux méthodes du génie génétique, il est effectivement possible modifier les « textes » de tous les génomes, soit en modifiant une lettre à la fois, soit en y insérant l’équivalent de « paragraphes » (au moins 300 lettres) qui coderaient pour une protéine.
Pour détecter si une insertion a eu lieu, il s’agit de procéder comme on le ferait pour détecter un plagiat dans un roman : on regarde s’il n’y a pas, par endroit, des similarités un peu trop flagrantes, par exemple un paragraphe quasi-identique dans les deux textes.
Si cette similarité concerne seulement quelques mots, ou une phrase (par exemple une citation), on estimera qu’il n’y a pas eu d’emprunt d’un texte dans un autre. Pour que le plagiat soit caractérisé, il faut que l’étendue cette ressemblance dépasse la longueur communément admise pour une coïncidence entre deux textes écrits dans la même langue.
Comment comparer des génomes ?
La comparaison de deux génomes (pour détecter les éventuels emprunts de gènes de l’un vers l’autre) repose sur le même principe. Comme les génomes sont écrits avec les mêmes lettres ATGC, la détection d’une suite de lettres similaires n’est le signe d’un emprunt (une manipulation génétique) que si celle-ci est plus longue que ce que l’on attend des similarités restreintes qui peuvent être liées au hasard dans deux textes écrits totalement indépendamment.
Des méthodes universellement acceptées permettent :
- d’identifier facilement les zones de plus grande ressemblance entre les génomes du Cov-2 et du VIH
- de démontrer que leurs niveaux de similarité ne dépassent pas ce que l’on attend du hasard, et donc qu’elles ne constituent pas la preuve d’un emprunt (c’est-à-dire d’une insertion ou manipulation effectuée par des chercheurs).
Détection des similarités par calcul statistique.
La plus forte similarité détectée par la comparaison des 30.000 lettres du CoV-2 avec les 9.200 du VIH est une « phrase » de 38 lettres, dont 33 sont identiques, au prix d’une insertion (« - »):
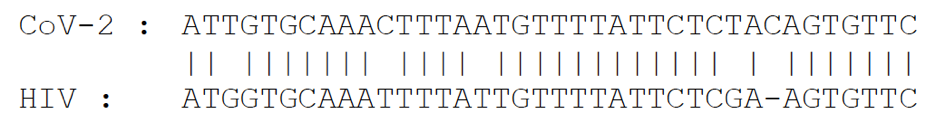
La seconde, moins bonne (28/30) est la suivante :
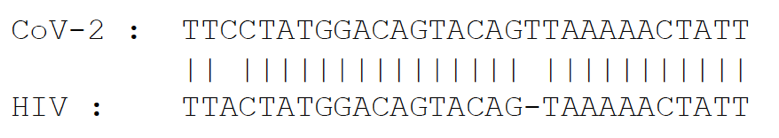
Un calcul (standard) des probabilités associé à ce type d’analyse nous indique que des zones atteignant ce niveau de similarité sont attendues plus de trois fois au hasard, c’est-à-dire en comparant des séquences de mêmes longueurs et de même composition en A,T,G et C fabriquées par un tirage aléatoire.
Conclusion : ces similarités ne sont en aucun cas inhabituelles, et ne peuvent pas servir d’argument en faveur d’une manipulation génétique qui aurait inséré un bout du génome VIH dans celui de SARS-CoV-2.
Mais une autre démonstration, qui ne fait pas appel à un calcul statistique, est encore plus probante.
Si l’on reprend maintenant le bout de séquence de SARS-CoV-2 qui aurait été emprunté au HIV (selon M. Montagnier) :
ATTGTGCAAACTTTAATGTTTTATTCTCTACAGTGTTC
et qu’on le cherche dans les textes des génomes d’autres souches de coronavirus bien plus anciennes (et naturellement associés aux chauves-souris), on peut vérifier qu’il est bien présent :
Exemple pour un virus isolé en 2005 (similarité 34/38):
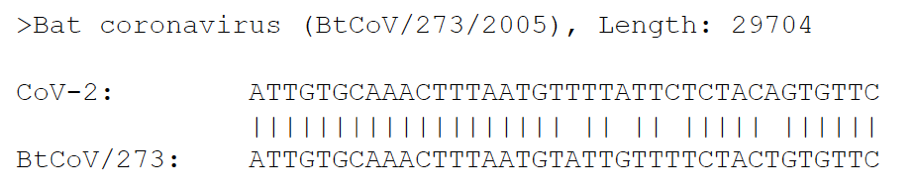
Cette zone de séquence code pour la séquence protéique « CANFNVLFSTVF » (les C, A, N …., lettres symbolisent différents acides aminés) conservée à l’identique dans toutes les souches de coronavirus car elle appartient à l’enzyme qui réplique le génome du virus (RNA polymerase) et dont la fonction lui est essentielle..
Conclusion, cette zone de ressemblance avec le VIH est fortuite, et existait dans la plupart des souches de coronavirus et ce, bien avant l’émergence de la Covid-19.
Dernier argument !
Enfin, si l’on se concentre sur la protéine du SARS-CoV-2 qui est la plus exposée à la surface du virus (la fameuse protéine « spike »), et qui serait donc la cible à privilégier pour faire un vaccin (car elle est la cible des anticorps) : sa comparaison détaillée avec la protéine d’enveloppe du virus VIH (la cible privilégiée pour faire un vaccin contre ce virus), à l’aide des méthodes universellement admises, ne détecte AUCUNE similarité.












































")



Commentaires
FredericN
20.04.2020 19h55FredericN
20.04.2020 22h47pourquoidocteur
21.04.2020 13h18Socrates
21.04.2020 13h37Socrates
21.04.2020 15h27FredericN
22.04.2020 10h48Merci pour vos éclaircissements qui sont très appréciés.
Je comprends tout à fait que le tirage statistique aléatoire avec 1 chance sur 4 n’est qu’une approximation, mais il a le mérite d’être facilement illustratif et c’est pourquoi je pense que c’est une bonne idée que vous l’ayez utilisé dans cet article.
A l’exception des nouvelles informations que vous indiquez (nombre de nucléotides à prendre en compte), il me semble que mon calcul initial tenait bien compte des points que vous indiquez dans votre réponse.
Si l’on considère le résultat 33/38, la première proba P1 était la chance de pouvoir trouver à l’identique une chaine de 38 lettres issue du HIV dans le Covid.
Cette proba allait ensuite être corrigé en P2 par le fait que 5 lettres étaient erronées, afin de donner le 33/38 que vous indiquez.
P1 peut se considérer comme la proba de tirer 38 lettres exactes d’affilé (1/4 exp 38, que je vais noter comme vous 1/4^38), avec autant de résultats acceptables que l’on peut découper le Covid en chaines de 38 lettres (on peut surestimer cela en supposant que ce nombre = nombre de nucléotides du Covid), et avec autant de tirages que l’on peut découper le VIH en chaines de 38 lettres (on peut surestimer cela en supposant que ce nombre = nombre de nucléotides du VIH).
P1 = (1/4^38) x Nbre nucleotide Covid x Nbre nucleotides HIV
P2 corrige P1 en tenant compte qu’il y a 5 lettres erronées. Ces dernières peuvent se trouver partout dans la séquences de 38 lettres mais sont interchangeables (C(38, 33) ≈ 5x10^5, meilleure notation que ce que j’indiquais par 38*37*36*35*34/(5*4*3*2)). Il faut d’autre part pour ces 5 lettres faire sauter la contrainte d’être exacte donc on multiplie par 4^5
Au final : P2 = P1 x C(38,33)x4^5 = (1/4^33) x 5x10^5 x Nbre nucleotide Covid x Nbre nucleotides HIV
En utilisant : Nbre nucleotide Covid = 30 000 et Nbre nucleotides HIV = 9200, on trouve : P2 = 1.9*10^-6, soit moins de 2 chances sur 1 million.
Si je prends les nouveaux chiffres que vous indiquez :
Nbre de nucléotides à prendre en compte sur SARS-CoV2 : 60 000 (et non 30 000), soit 2 fois plus.
Nbre de nucléotides à prendre en compte sur HIV : 100 000 (et non 9200), soit 10 fois plus
On arrive à P’2 = P2 x 20 = 4 chances sur 100 000, et cela pour la seule séquence 33/38.
Du fait que l’on prend en compte 100 000 nucléotides pour le HIV, il me semble que l’on peut multiplier les proba 33/38 et 28/30, ce qui diminue encore cette proba par à peu près le même nombre ?
Merci de vos retours.